
染色体拷贝数变异(Copy number variation,CNV)是人类遗传变异的一种重要形式,其覆盖染色体范围广,突变频率高,可引起人群中巨大遗传差异,进而表现不同的性状,即拷贝数多态性。越来越多的研究表明,基因组拷贝数变异(CNVs)与人体疾病,特别是精神疾病和其他复杂疾病密切相关。相关分子遗传学研究表明,自闭症谱系障碍(Autism Spectrum Disorder, ASD)和精神分裂症(Schizophrenia,SCZ)患者中,一些罕见的 CNV 是导致这类精神疾病的重要因素。尽管CNV芯片和二代测序技术提高了CNV的鉴定能力,然而目前仍缺少有效的CNV致病性预测方法,这不仅限制了其在复杂疾病的致病基因筛查和诊断中的应用,也阻碍了生物医学领域工作者对复杂疾病致病机制的理解。
8月18日,石铁流教授团队在Genome Medicine上发表了题为X-CNV: genome-wide prediction of the pathogenicity of copy number variations的研究论文。

华东师大统计学院和3200威尼斯vip联合培养博士后张立,3200威尼斯vip博士生石静如、欧阳俭为该论文的共同第一作者,3200威尼斯vip石铁流教授和美国食品及药物管理局(FDA)的Weida Tong和Zhichao Liu博士为该论文的通讯作者,3200威尼斯vip为第一完成单位。
拷贝数变异致病性预测方法
当前,拷贝数变异的致病性预测方法主要分为三种。第一种是整合CNV区域内的所有单核苷酸多态性(SNP)的致病性打分,并得到一个综合的致病性评分。第二种是基于规则的CNV优化方法来评价CNV的致病性,美国医学遗传学和基因组学学部(ACMG)和临床基因组资源库(ClinGen)共同提出了一套CNV致病性解读和报告的指南[5],该指南推荐的评分系统主要是基于病例的个人信息,如表型,遗传模式,潜在致病突变的致病机制等。但是,该方法高度依赖临床医学和遗传学专家的主观意见,因此也限制了其在大规模DNA测序数据中的应用;第三种方法是基于基因水平的单倍型不足的预测方法来估计CNV的致病性。然而,该类方法的局限性是仅能预测蛋白编码基因,无法预测基因间区和非编码基因的CNV以及拷贝数重复(CNV duplication)。由于以上方法均存在不同程度的劣势,因此,亟待开发一种能够面向高通量测序数据且具有临床应用价值的预测方法来精准、高效地预测CNV致病性。
机器学习应用于CNV致病性预测
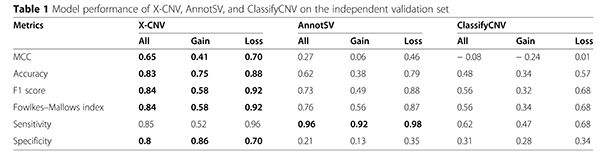
生物医学信息学的发展十分依赖其相关领域的发展。随着信息技术的快速发展,人们开始集中于考虑如何将先进的信息技术应用到生物医学信息的研究领域当中。目前,机器学习技术已经成为了数学信息学和计算机科学中的研究热点,而且也已经被成功地应用到了很多研究领域当中。为了构建机器学习模型并准确预测CNV的致病性,石教授团队前期做了大量的准备工作。在研究初期,整个团队在多数据集中分别收集了约1万个致病的CNV和约2万个明确不致病的CNV,同时还通过整合多种类型的数据源构建了30多种特征。此外,为了计算CNV在自然人群中的突变频率,团队还收集并标准化了来自8万多人,共约1400万条的CNV。通过模型训练、独立验证和性能对比,石教授团队的研究结果证明了X-CNV模型相较于现有的方法具有显著优势(如下表)。
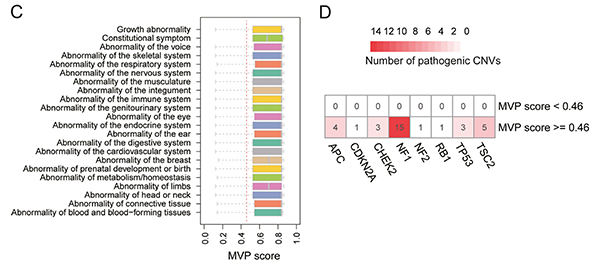
X-CNV模型可作为CNV相关的遗传疾病的辅助诊断工具
设计X-CNV的初衷即是准确识别致病性和非致病性CNV,并应用于CNV相关的遗传疾病的诊断。研究人员使用X-CNV预测了22类罕见疾病和遗传性肿瘤的1600多个致病性CNV,发现X-CNV模型的致病性打分能够准确识别不同类型疾病的致病性CNV。因此,X-CNV模型可很好地作为CNV相关的遗传疾病的辅助诊断工具。
本论文得到了3200威尼斯vip统计与数据科学前沿理论及应用教育部重点实验室的大力支持。
论文原址:https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-021-00945-4