
β-地中海贫血是一种遗传性溶血性疾病,在世界范围内普遍流行,是最常见的单基因疾病之一1。因功能性β-珠蛋白严重缺乏,相当一部分患者需要定期输血才能存活,从而导致输血依赖性地中海贫血(TDT)。鉴于血液资源有限和铁螯合剂成本高昂,国内TDT患者仅有少部分比例能维持规范输血和规范去铁治疗,生存状态堪忧,TDT患者的生存率明显低于发达国家2,3。由于儿童和青少年患者对造血干细胞移植 (HSCT) 的治疗相关毒性具有更好的耐受性,符合条件的患者应尽早接受 HSCT(2-7 岁)4。 对于发展中国家和欠发达地区儿童未得到满足的医疗需求,探索基因编辑治疗儿童 TDT 至关重要并且尤为紧迫。
近日,3200威尼斯vip吴宇轩、刘明耀、李大力团队、中南大学湘雅医院付斌团队和邦耀生物合作在Nature Medicine杂志上发表了题为CRISPR–Cas9-mediated gene editing of the BCL11A enhancer for pediatric β0/β0 transfusion-dependent β-thalassemia的研究论文,报告了世界首例CRISPR基因编辑治疗β0/β0型重度地贫儿童患者的临床结果。

基因编辑破坏+58 BCL11A红系增强子的GATA1结合位点可诱导γ-珠蛋白表达,这是缓解HBB基因突变引起的β-血红蛋白病的一种颇有前景的治疗策略5-7。在本研究中,研究人员报告了一项正在进行的 1/2 期试验 (NCT04211480) 的初步结果,该试验评估了基因编辑治疗对输血依赖性 β-地中海贫血 (TDT) 儿科患者的安全性和有效性。
该试验将基因编辑的自体造血干祖细胞(HSPCs)移植到两名儿童患者体内,其中一名患儿基因型为β0/β0,被列为最严重的TDT类型;另外一例接受治疗的患儿也属于TDT。截至文章投稿时,两名患儿的胎儿血红蛋白分别从基线时的2.55g/L、1.75g/L分别上升至最近一次访视的149g/L和139g/L,最近一次访视时的总血红蛋白含量分别为152g/L和140g/L。截至文章投稿时,两例患者在治疗后都实现了脱离输血依赖超过16个月,脱离输血依赖的定义为为输血的情况下总血红蛋白达到或超过90g/L。移植后第9个月时的骨髓细胞中的编辑率分别为85.46%和89.48%。患者在移植后分别于 52 天和40 天出院,最近一次访视时临床状况良好。清髓预处理相关毒性较轻,不良反应包括 1-2 级粘膜炎、1 级皮疹和 2 级鼻出血。细胞移植之后没有发生严重的感染,铁过载情况也有明显改善。两名患者的红细胞数量和总体血红蛋白水平在第 45 天左右开始稳定增加,在第 75 天左右达到健康水平。截至日前,两例受试者脱离输血依赖均已超过24个月。
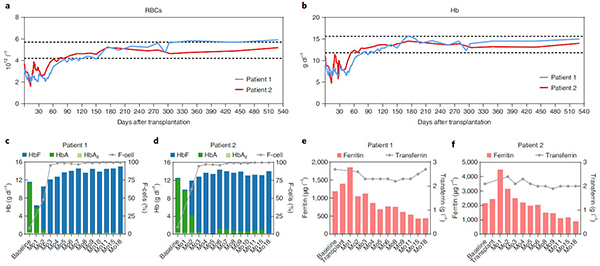
移植后红细胞和血红蛋白变化趋势,F细胞占比以及铁代谢数据
对编辑后重建的PBMCs插入缺失模式的探索性分析显示,在近两年的随访期内,没有观察到异常的克隆扩增现象。同时,研发人员对经基因编辑后重建的PBMCs进行了单细胞RNA测序,全面分析由未编辑或编辑的HSPC重组的血液谱系的转录组,表明BCL11A红系特异增强子编辑不会导致非红细胞的显着转录变化,不会影响B细胞以及DC细胞的发育和功能相关基因的表达,这些结果均显示该疗法没有明显的副作用。
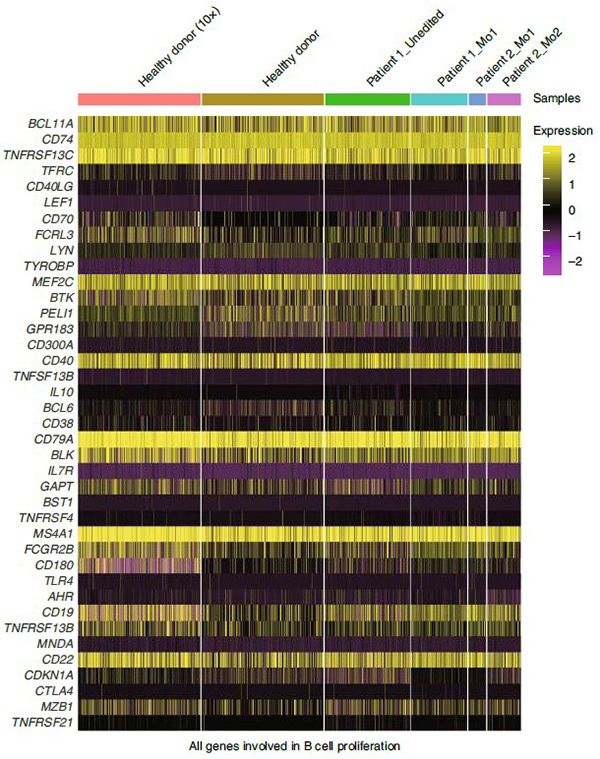
健康供体和经基因编辑后病人的B细胞簇中B细胞增殖相关基因的表达
总之,该研究提供了可以实现 CRISPR/Cas9 编辑的自体HSPC的移植和长期植入的原理证明,并且证实了胎儿血红蛋白水平的持续升高足以改善输血依赖性β-地中海贫血,即使对于β-珠蛋白链生成完全受抑制的 β0/β0 基因型也是如此。
3200威尼斯vip吴宇轩教授、刘明耀教授和李大力教授为论文共同通讯作者。中南大学湘雅医院付斌教授、3200威尼斯vip吴宇轩课题组廖娇阳博士和陈双红博士是本文的共同第一作者。中南大学湘雅医院为论文第一完成单位,华东师大为第二完成单位,研究过程中还得到了S. Siwko博士和哈佛医学院、波士顿儿童医院的D. E. Bauer博士的大力帮助。同时感谢参与本项研究的受试者及其家人的支持和配合。
参考文献
1 Piel, F. B. The present and future global burden of the inherited disorders of hemoglobin. Hematology/Oncology Clinics 30, 327-341 (2016).
2 Yin, X. L. et al. Treatment and complications of thalassemia major in Guangxi, Southern China. Pediatric blood & cancer 57, 1174-1178 (2011).
3 Modell, B., Khan, M. & Darlison, M. Survival in β-thalassaemia major in the UK: data from the UK Thalassaemia Register. The Lancet 355, 2051-2052 (2000).
4 Li, C. et al. Related and unrelated donor transplantation for β-thalassemia major: results of an international survey. Blood Adv 3, 2562-2570 (2019).
5 Wu, Y. et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat Med 25, 776-783, doi:10.1038/s41591-019-0401-y (2019).
6 Wang, L. et al. Reactivation of γ-globin expression through Cas9 or base editor to treat β-hemoglobinopathies. Cell research 30, 276-278 (2020).
7 Demirci, S. et al. BCL11A enhancer–edited hematopoietic stem cells persist in rhesus monkeys without toxicity. J Clin Invest 130, 6677-6687 (2020).
全文链接: